Introduction
iProPhos is a user-friendly interactive web portal that provides multiple analysis modules to explore and visualize functional proteomics and phosphoproteomics across 12 cancer types.
Data Source
iProPhos contains a large number of samples including 1,444 tumor samples and 746 normal samples across 12 cancer types. Transcriptome, proteome, phosphoproteome, and clinical data are obtained from the Clinical Proteomic Tumor Analysis Consortium (CPTAC) project (https://proteomics.cancer.gov/programs/cptac).
| CPTAC | Disease Type | Proteome (Tumor+Normal) | Phosphoproteome (Tumor+Normal) | Transcriptome (Tumor only) | Publication |
|---|---|---|---|---|---|
| BRCA | Breast Invasive Carcinoma | 140(122+18) | 140(122+18) | 122 | PMID: 33212010 |
| CCRCC | Clear Cell Renal Cell Carcinoma | 194(110+84) | 194(110+84) | 110 | PMID: 31675502 |
| COAD | Colon Adenocarcinoma | 197(97+100) | 197(97+100) | 96 | PMID: 31031003 |
| GBM | Glioblastoma | 109(99+10) | 109(99+10) | 99 | PMID: 33577785 |
| HCC | HBV-Related Hepatocellular Carcinoma | 318(159+159) | 318(159+159) | 159 | PMID: 31585088 |
| HNSCC | Head and Neck Squamous Cell Carcinoma | 171(108+63) | 171(108+63) | 108 | PMID: 33417831 |
| LUAD | Lung Adenocarcinoma | 211(110+101) | 211(110+101) | 110 | PMID: 32649874 |
| LSCC | Lung Squamous Cell Carcinoma | 207(108+99) | 207(108+99) | 108 | PMID: 34358469 |
| OV | Ovarian Serous Cystadenocarcinoma | 103(83+20) | 103(83+20) | 82 | PMID: 32529193 |
| PBT | Pediatric Brain Tumor | 218(218+0) | 218(218+0) | 188 | PMID: 33242424 |
| PDA | Pancreatic Ductal Adenocarcinoma | 202(135+67) | 202(135+67) | 135 | PMID: 34534465 |
| UCEC | Uterine Corpus Endometrial Carcinoma | 120(95+25) | 120(95+25) | 95 | PMID: 32059776 |
[For plot]
Differential Analysis Results
Differential analysis is conducted employing the
limma
algorithm.
Download table
Proteins for GO Enrichment Input
The table below displays the results of differential analysis conducted using the limma algorithm. The data has been filtered based on your customized cutoff values. Please review the listed proteins for further input in the GO enrichment analysis. Then, click on the
"Plot"
button to generate plots.
GO graph
Proteins for KEGG Enrichment Input
The table below displays the results of differential analysis conducted using the limma algorithm. The data has been filtered based on your customized cutoff values. Please review the listed proteins for further input in the KEGG enrichment analysis. Then, click on the
"Plot"
button to generate plots.
KEGG graph
Proteins for PPI Input
The table below displays the results of differential analysis conducted using the limma algorithm. The data has been filtered based on your customized cutoff values. Please review the listed proteins for further input in the PPI analysis. Then, click on the
"Plot"
button to generate plots.
iProPhos supports the visualization of a PPI network for up to 200 differential proteins, ranked by logFC.
PPI graph
[For plot]
Differential Analysis Results
Differential analysis is conducted employing the
limma
algorithm.
Download table
File upload
Data format:
Both
txt
and
csv
formats of files are acceptable.
Proteomics data
Please upload the file format like this:
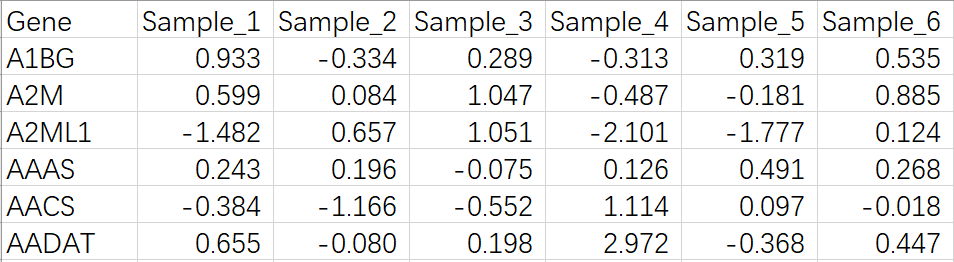
For the first column, please input
gene symbols
of proteins.
Phosphoproteomics data
Please upload the file format like this:
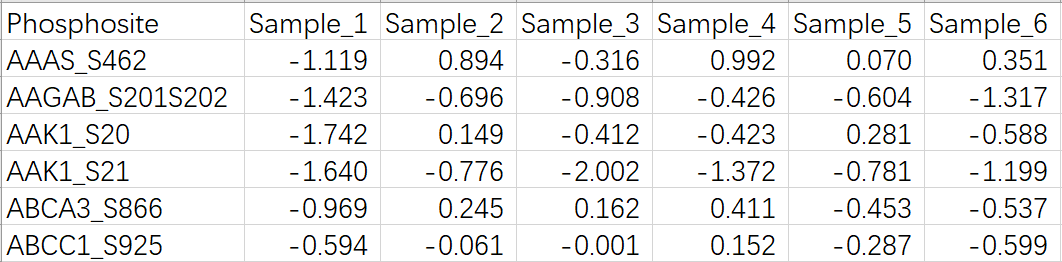
For the first column, please input
gene symbol_phosphosites
.
Note:
The size of the uploaded file should not exceed
60M
.
iProPhos will not process the uploaded proteomics/phosphoproteomics data, and NA values will be filtered out during the analysis.
Data format:
Both
txt
and
csv
formats of files are acceptable.
Sample group
Please upload the file format like this:

-
case_id: Keep the same number and sample names as those in the proteomics/phosphoproteomics data.
-
class: Sample classification, including two categories:
Tumor/Normal. If there is no normal group, you should fill inTumorfor all samples.
Data format:
Both
txt
and
csv
formats of files are acceptable.
Survival information
Please upload the file format like this:
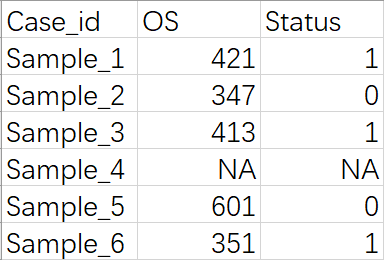
-
Case_id: Match the "case_id" of
tumor samplesin the sample group file. -
OS: Overall survival time. It refers to the length of time from a specific event (such as diagnosis or treatment initiation) until the patient's death from any cause or the end of the study period.
-
Status: Patient's survival status. Status 1 = dead, 0 = alive or censored.
If the patient's survival information is missing, fill it with
NA
.
The Example data could be downloaded here.
Differential Analysis Results
Differential analysis is conducted employing the
limma
algorithm.
Download table
GO enrichment
KEGG enrichment
GSEA
The protein list used for GSEA is ranked based on log2(fold change) from differential expression analysis using the limma algorithm.
Overview
iProPhos can perform proteomics-related and phosphoproteomics-related analyses.
Proteome Analysis
Differential analysis
This feature allows users to explore and compare the expression patterns of their interested proteins across tumor and normal samples.
Boxplot
iProPhos generates boxplots with jitter and allows users to customize box color, point size and statistical methods.
Parameters
-
Dataset: Select a cancer type of interest.
-
Protein: Input a protein of interest.
Note:The available proteins in each dataset vary. Only 1000 proteins from the respective dataset are shown in the dropdown list, and users can also manually input proteins with auto-completion. If a protein that is not present in the selected dataset is input, it will be treated as a null value and result in an error message. -
Tumor color: Set the box color in tumor samples.
-
Normal color: Set the box color in normal samples.
-
Point Size: Set the point size.
-
Differential Methods: Select a method for differential analysis.
-
t-test: two-tailed test, assuming unequal variances.
-
wilcox.test: Wilcoxon rank-sum test.
-
anova: assuming equal variances.
The t-test is appropriate when the data is normally distributed. The Wilcoxon test is suitable when the data does not meet the assumptions of normality. ANOVA is useful when assuming normality and equal variances. The choice of the appropriate test should be based on the specific characteristics of the data.
This analysis involves individual protein without multiple comparisons, so it is not corrected for multiple testing.
-
Results

Volcano plot
iProPhos generates volcano plots and allows users to set the cutoff value to define significance.
Parameters
- Dataset: Select a cancer type of interest.
[For plot]
- Protein: Input a protein of interest.
- FDR cutoff: Input the adjusted p-value cutoff.
- |log2FC| cutoff: Input the |log2(fold change)| cutoff. This value should be greater than 0.
Results
Plot
Upregulated and downregulated proteins in tumor samples are labeled orange and blue respectively, while gray means non-significance. Moreover, the interested protein can be magnified and highlighted with its gene symbol.

Table
This table (ranked by |logFC|) provides a concise summary of the differential analysis results using the limma algorithm.

Correlation analysis
iProPhos allows users to evaluate protein expression correlations with scatter plots or tables.
Correlation plot
This feature investigates the correlation between two interested proteins in the specific tumor.
Parameters
-
Dataset: Select a cancer type of interest.
-
Protein A: Input a protein A of interest. [For x-axis]
-
Protein B: Input a protein B of interest. [For y-axis]
-
Color for non-imputed data: Set the point color for non-imputed data.
-
Color for imputed data: Set the point color for KNN imputed data.
-
Point Size: Set the point size.
-
Method: Select a method for the correlation test.
-
pearson: Pearson correlation assumes that the variables are normally distributed and have a linear relationship.
-
spearman: Spearman correlation assesses the non-linear relationship between two variables, and it does not assume normality.
-
kendall: Kendall correlation is a non-parametric correlation measure that assesses the strength of association between two variables without assuming linearity.
This analysis involves individual protein pair without multiple comparisons, so it is not corrected for multiple testing.
-
Results

Correlation table
This table shows the correlation of the target protein with other proteins in the selected dataset.
Parameters
-
Dataset: Select a cancer type of interest.
-
Protein: Input a protein of interest.
-
Method: Select a method for the correlation test.
Results
Correlation analysis provides results both with and without imputation. The table has been ranked by the correlation coefficient, and p-values have been adjusted using the Benjamini-Hochberg (BH) method.
Using non-imputed dataset

Using imputed dataset
The missing values have been imputed using KNN algorithm.

Survival
This feature enables the identification of potential biomarkers which significantly associate with clinical outcome. iProPhos performs overall survival (OS) analysis based on protein abundance. Log-rank test has been used for hypothesis test.
Parameter
-
Dataset: Select a cancer type of interest.
-
Protein: Input a protein of interest.
-
“Group: High” color: Choose the color for the high-expression group.
-
“Group: Low” color: Choose the color for the low-expression group.
-
Group Cutoff: Select an appropriate expression threshold to divide patients into high-expression and low-expression groups. Median means the median value of protein abundance, and optimal value is determined by the surv_cutpoint algorithm, which exhibits the most significant association with survival.
This analysis involves individual protein without multiple comparisons, so it is not corrected for multiple testing.
Results

mRNA&Protein Correlation
iProPhos performs correlation analysis between mRNA and protein abundances in specific cancer type.
Parameters
-
Dataset: Select a cancer type of interest.
-
Gene: Input a gene of interest.
-
Color for non-imputed data: Set the point color for non-imputed data.
-
Color for imputed data: Set the point color for KNN imputed data.
-
Point Size: Set the point size.
-
Method: Select a method for the correlation test.
-
pearson: Pearson correlation assumes that the variables are normally distributed and have a linear relationship.
-
spearman: Spearman correlation assesses the non-linear relationship between two variables, and it does not assume normality.
-
kendall: Kendall correlation is a non-parametric correlation measure that assesses the strength of association between two variables without assuming linearity.
This analysis involves individual mRNA-protein pair without multiple comparisons, so it is not corrected for multiple testing.
-
Results

Clinical
iProPhos investigates the association between protein abundance and clinical features, such as age, gender and tumor stage.
Age
Parameters
-
Dataset: Select a cancer type of interest. The age ranges of patients in the specific datasets have been labeled.
-
Protein: Input a protein of interest.
-
Younger color: Set the violin color for young patients.
-
Older color: Set the violin color for old patients.
-
Method: Choose an appropriate method to classify patients into young and old groups. Median means the median age of patients. When users select custom method, they can input a suitable number as the cutoff value.
-
Differential Methods: Select a method for differential analysis.
-
t-test: two-tailed test, assuming unequal variances.
-
wilcox.test: Wilcoxon rank-sum test.
-
anova: assuming equal variances.
The t-test is appropriate when the data is normally distributed. The Wilcoxon test is suitable when the data does not meet the assumptions of normality. ANOVA is useful when assuming normality and equal variances. The choice of the appropriate test should be based on the specific characteristics of the data.
This analysis involves individual protein without multiple comparisons, so it is not corrected for multiple testing.
-
Results

Gender
-
Dataset: Select a cancer type of interest.
-
Protein: Input a protein of interest.
-
Female color: Set the violin color for female patients.
-
Male color: Set the violin color for male patients.
-
Differential Methods: Select a method for differential analysis.
This analysis involves individual protein without multiple comparisons, so it is not corrected for multiple testing.
Results

Tumor stage
-
Protein: Input a protein of interest.
-
Dataset: Select a cancer type of interest.
The method for differential analysis is one-way ANOVA, using tumor stage as variable for calculating protein differential expression.
This analysis involves individual protein without multiple comparisons, so it is not corrected for multiple testing.
Results

Enrichment
iProPhos performs over-representation analysis (ORA) and gene set enrichment analysis (GSEA) to identify dysregulated pathways. The enrichment analyses are based on the results of differential expression analysis using the limma algorithm. ORA analysis includes Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses.
GO enrichment
Parameter
- Dataset: Select a cancer type of interest.
- Regulation: Select the up-regulated or down-regulated proteins in the tumor.
- adjusted P-value cutoff: Set the cutoff value for the adjusted p-value (BH correction).
- Fold change cutoff: Set the cutoff value for the fold change (tumor vs normal).
Results

The redundant GO terms are removed to present the most informative terms with simplify algorithm.
The graphic results show the top10 enriched annotations for biological process (BP), cellular component (CC) and molecular function (MF). iProPhos also provides a complete list of GO enrichment results for download.
KEGG enrichment
Parameter
- Dataset: Select a cancer type of interest.
- Regulation: Select the up-regulated or down-regulated proteins in the tumor.
- adjusted P-value cutoff: Set the cutoff value for the adjusted p-value (BH correction).
- Fold change cutoff: Set the cutoff value for the fold change (tumor vs normal).
Results

The graphic results show the top10 enriched KEGG annotations. iProPhos also provides a complete list of KEGG enrichment results for download.
GSEA
Parameter
- Gene list: Input the gene list of interest, separated by commas.
- Dataset: Select a cancer type of interest.
Results

PPI
iProPhos generates an interactive network diagram of protein interactions (including both physical and functional interactions) to performs the protein-protein interaction (PPI) network analysis. The PPI network analysis is based on the results of differential expression analysis using the limma algorithm.
Parameter
- Dataset: Select a cancer type of interest.
- Regulation: Select the up-regulated or down-regulated proteins in the tumor.
- adjusted P-value cutoff: Set the cutoff value for the adjusted p-value (BH correction).
- Fold change cutoff: Set the cutoff value for the fold change (tumor vs normal).
Results

Nodes represent proteins, and edges represent their interactions, with thicker edges indicating higher confidence of interactions. Users can download the results in a tabular format for further analysis in Cytoscape or other similar software.
Phosphoproteome Analysis
This module aims to do phosphoproteomics-related analysis and integrate with proteomics data. Differential analysis and clinical features-related analysis are the same as those in the Proteome Analysis module. Therefore, these features will not be repeated here.
Note: The available phosphosites in each dataset vary. Only 1000 phosphosites from the respective dataset are shown in the dropdown list, and users can also manually input phosphosites with auto-completion. If a phosphosite that is not present in the selected dataset is input, it will be treated as a null value and result in an error message.
Correlation
This feature investigates correlations between protein abundance and the phosphorylation levels of phosphosites.
Parameter
-
Dataset: Select a cancer type of interest.
-
Protein: Input a protein of interest.
-
Site: Input the phosphosite of interest.
-
Color for non-imputed data: Set the point color for non-imputed data.
-
Color for imputed data: Set the point color for KNN imputed data.
-
Point Size: Set the point size.
-
Method: Select a method for the correlation test.
-
pearson: Pearson correlation assumes that the variables are normally distributed and have a linear relationship.
-
spearman: Spearman correlation assesses the non-linear relationship between two variables, and it does not assume normality.
-
kendall: Kendall correlation is a non-parametric correlation measure that assesses the strength of association between two variables without assuming linearity.
This analysis involves individual protein-phosphosite pair without multiple comparisons, so it is not corrected for multiple testing.
-
Results

Kinase-substrate Correlation
This feature investigates correlations between protein abundance of kinases and phosphorylation level of substrates. Results have been ordered by correlation coefficient.
Parameter
-
Dataset: Select a cancer type of interest.
-
Kinase: Select one kinase of interest.
The list of kinases was collected from the PhosphoSitePlus (http://www.phosphosite.org) and NetworKIN (https://networkin.info/).
-
**Method: **Select a method for the correlation test.
Results
Results are provided both with and without imputation. The table has been ranked by the correlation coefficient, and p-values have been adjusted using the BH correction.
Using non-imputed dataset

Using imputed dataset
The missing values have been imputed using KNN algorithm.

Kinase-Substrate Enrichment
iProPhos estimates changes in a kinase’s activity based on the collective phosphorylation changes of its identified substrates using kinase-substrate enrichment analysis (KSEA) algorithm. The following annotations refer to Wiredja D.D. et al publication.
Parameter
-
Dataset: Select a cancer type of interest.
-
Kinase-substrate dataset: Choose the data sources of kinase-substrate relationship. PhosphoSitePlus: PhosphoSitePlus only includes experimentally verified kinase-substrate relationships, which is recommended for the more conservative results. PhosphoSitePlus + NetworKIN: NetworKIN provides predicted relationships. Users can choose this option to score more kinases. Once selected, users should set a NetworKIN score cutoff, which indicates that higher scores have more confident kinase and substrate prediction.
-
[for plot] p-value cutoff: Set p-value cutoff to mark the statistically significant kinases in the bar plot. Red means significantly positive scores and blue means significantly negative scores, whereas black means insignificant. The value of scores implies the overall change of kinase activity relative to normal samples.
-
[for plot] Substrate count cutoff: Set the minimal substrates which correspond to a kinase in the bar plot. A lower cutoff thus allows more kinases into the bar plot.
Results
Kinase Barplot
Show the kinase scores in the form of bar plot. Note: not all kinases in this bar plot, it is decided by the substrate count cutoff option.

Kinase scores
This table lists all kinases which have at least one identified substrate in the selected dataset, thus also includes kinases not marked in the bar plot. Please refer to the original Casado et al. publication for detailed description what these columns represent.
-
Kinase.Gene: the gene name of each kinase.
-
mS: the mean log2(fold change) of all the kinase’s substrates.
-
Enrichment: the background-adjusted value of the kinase’s mS.
-
m: the total amount of detected substrates from the kinase-substrate dataset for each kinase.
-
z.score: the normalized score for each kinase, weighted by the number of identified substrates.
-
p.value: the statistical assessment for the z.score.
-
FDR: the p-value adjusted for multiple hypothesis testing using the BH method.

Kinase-Substrate Link
This table lists all the kinase and substrate relationships identified from the selected dataset. It also includes relationships for kinases not marked in the bar plot.
-
Kinase.Gene: the gene name for each kinase.
-
Substrate.Gene: the gene name for each substrate linked to that kinase.
-
Substrate.Mod: the substrate’s specific amino acid residue that was modified.
-
Source: the database where the kinase-substrate annotation was derived from.
-
log2FC: the log2(fold change) value of that particular phosphosite.

Survival
This feature can find the potential phosphosites significantly correlated with overall survival (OS). Log-rank test has been used for hypothesis test.
Survival plot
This feature provides OS analysis of one interested phosphosite with the Kaplan-Meier survival curve plot.
Parameter
-
Dataset: Select a cancer type of interest.
-
Phosphosite: Input one phosphosite of interest.
-
“Group: High” color: Choose the color for the group with high phosphorylation levels.
-
“Group: Low” color: Choose the color for the group with low phosphorylation levels.
-
Group Cutoff: Choose an appropriate threshold of phosphorylation levels to separate patients into the groups with high and low phosphorylation levels. Median means the median value of phosphorylation levels, and optimal value is determined by the
surv_cutpointalgorithm that exhibits the most significant association with survival.This analysis involves individual phosphosite without multiple comparisons, so it is not corrected for multiple testing.
Results

Survival table
This feature provides OS analysis of all detected phosphosites in a given protein in the selected dataset. With this function, users could easily select the phosphosites that significantly correlated with survival outcomes for further research. This feature uses the median value of phosphorylation levels as the cutoff to categorize patients into high and low phosphorylation level groups.
Parameter
-
Dataset: Select a cancer type of interest.
-
Protein: Input the protein of interest.
Result

User upload
users can upload their own proteomics/phosphoproteomics data for analysis and visualization.
File upload
[Required]
-
proteomics/phosphoproteomics data
-
sample group imformation
[Optional]
- patient survival information
Data format:
Both txt and csv formats of files are acceptable.
Proteomics data
Please upload the file format like this:

For the first column, please input gene symbols of proteins.
Phosphoproteomics data
Please upload the file format like this:

For the first column, please input gene symbol_phosphosites .
Note:
The size of the uploaded file should not exceed 60M .
iProPhos will not process the uploaded proteomics/phosphoproteomics data, and NA values will be filtered out during the analysis.
Sample group
Please upload the file format like this:

-
case_id: Keep the same number and sample names as those in the proteomics/phosphoproteomics data.
-
class: Sample classification, including two categories:
Tumor/Normal. If there is no normal group, you should fill inTumorfor all samples.
Survival information
Please upload the file format like this:

- Case_id: Match the “case_id” of
tumor samplesin the sample group file. - OS: Overall survival time. It refers to the length of time from a specific event (such as diagnosis or treatment initiation) until the patient’s death from any cause or the end of the study period.
- Status: Patient’s survival status. Status 1 = dead, 0 = alive or censored.
If the patient’s survival information is missing, fill it with NA .
 Download
DownloadProteome, phosphoproteome, and clinical data are available for download across 12 cancer types.
| CPTAC | Disease Type | Download |
|---|---|---|
| BRCA | Breast Invasive Carcinoma | BRCA.zip |
| CCRCC | Clear Cell Renal Cell Carcinoma | CCRCC.zip |
| COAD | Colon Adenocarcinoma | COAD.zip |
| GBM | Glioblastoma | GBM.zip |
| HCC | HBV-Related Hepatocellular Carcinoma | HCC.zip |
| HNSCC | Head and Neck Squamous Cell Carcinoma | HNSCC.zip |
| LUAD | Lung Adenocarcinoma | LUAD.zip |
| LSCC | Lung Squamous Cell Carcinoma | LSCC.zip |
| OV | Ovarian Serous Cystadenocarcinoma | OV.zip |
| PBT | Pediatric brain tumor | PBT.zip |
| PDA | Pancreatic Ductal Adenocarcinoma | PDA.zip |
| UCEC | Uterine Corpus Endometrial Carcinoma | UCEC.zip |
iProPhos
iProPhos is a user-friendly interactive web portal that provides multiple analysis modules to explore and visualize functional proteomics and phosphoproteomics across 12 cancer types.
iProPhos has “Proteome Analysis” and “Phosphoproteome Analysis” modules. In iProPhos, users can perform profiling plotting and differential expression, patient survival, clinical feature-related, and correlation analyses, including protein-protein, mRNA-protein, and kinase-substrate correlations. Furthermore, functional enrichment, protein-protein interaction network, and kinase-substrate enrichment analyses are accessible. And also, it is convenient for users to customize the analytic parameters, graphic color, and point size.
This tool is developed by Jing Zou, Ziran Qin and Ran Li of Zhang Lab, Zhejiang University.
Contact us
If you have any questions about iProPhos, please contact us: l_zhang@zju.edu.cn.
Acknowledgement
Transcriptome, proteome, phosphoproteome, and clinical data across 12 cancer types are obtained from the Clinical Proteomic Tumor Analysis Consortium (CPTAC) project (https://proteomics.cancer.gov/programs/cptac), which are computed by a standard pipeline.